
Диагностика СМА
При подозрении на СМА выбор методов обследования должен определяться анамнестическими данными и результатами клинического осмотра.
СМА следует подозревать во всех случаях тяжелой мышечной гипотонии у детей, нередко проявляющейся «синдромом вялого ребенка» и (или) характерной "позой лягушки" вследствие гипотонии проксимальных групп мышц.
Схожая клиническая картина может наблюдаться при врожденных миопатиях, мышечных дистрофиях, полиневропатиях и заболеваниях с поражением нервно-мышечного аппарата, с которыми следует дифференцировать СМА (особенно при подозрении на более легкие формы).1
Диагностический алгоритм включает проведение биохимического анализа крови с исследованием активности фермента креатининфосфокиназы (КФК). Известно, что активность креатинфосфокиназы в сыворотке крови у пациентов с СМА может превышать норму в 2–4 раза, но не более чем в 10 раз.2
Среди электрофизиологических методов диагностики используется метод игольчатой электромиографии (ЭМГ), которая позволяет исследовать конкретную мышцу и определить степень денервационной активности. При игольчатой ЭМГ характерно увеличение амплитуды интерференционной кривой, разреженность интерференционной кривой, появления различных потенциалов спонтанной активности, складывающихся в так называемый «ритм частокола».3

Общий алгоритм дифференциальной диагностики СМА5
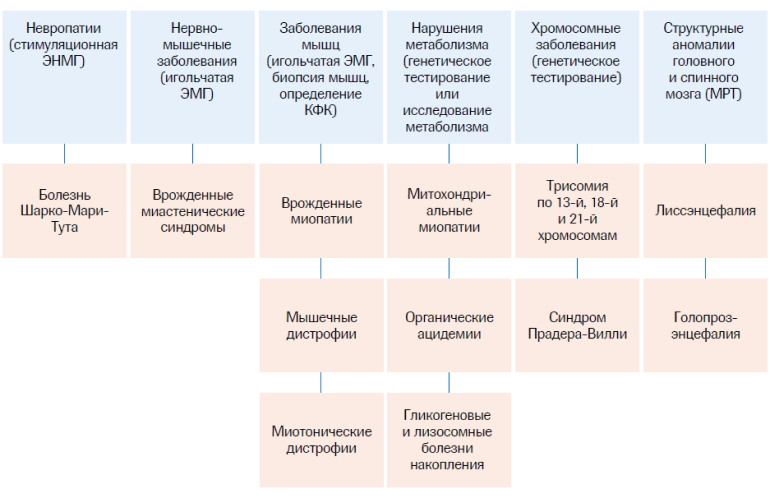
КФК – креатинфосфокиназа; МРТ – магнитно-резонансная томография; ЭМГ – электромиография; ЭНМГ – электронейромиография.
Генетическое тестирование
СМА вызывается мутациями в гене SMN1, кодирующем белок выживаемости мотонейронов (SMN). Именно идентификация гена SMN1 открыла возможность для разработки прямой ДНК-диагностики. Выявление мажорной мутации делеции экзонов 7 и/или 8 гена SMN1 является качественным, надежным и чувствительным диагностическим тестом - «золотым стандартом» диагностики СМА.
Наиболее частым генотипом СМА (до 95% случаев) является делеция в 7–8-м экзоне гена SMN1 в гомозиготном состоянии.14 Иногда СМА может развиваться в результате конверсии, при которой ген SMN1 становится похож по структуре на ген SMN2. Это приводит к двум функционирующим копиям гена SMN2 и отсутствию рабочих копий SMN1. Еще одной причиной СМА может являться компаунд-гетерозиготность в виде единичной делеции в одном аллеле SMN1 и иной мутации во втором аллеле.
Первым шагом в ДНК-диагностике СМА является определение гомозиготной делеции в SMN1. Дальнейшее обследование может включать в себя определение дозы гена SMN1 и его секвенирование с целью выявления компаунд-гетерозиготного состояния. Специфичность определения делеции близка к 100%, а чувствительность составляет 95%.4
Диагностический алгоритм для СМА
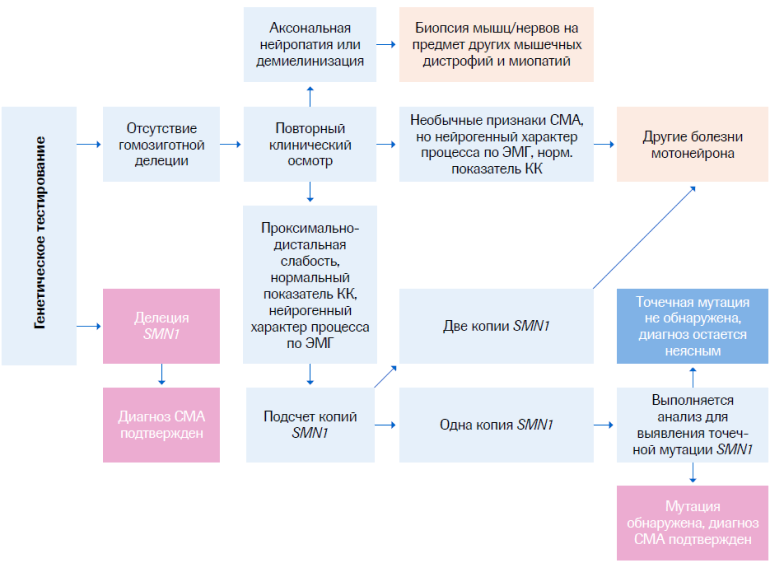
КК - креатинкиназа; ЭМГ - электромиография; ген SMN - ген выживаемости мотонейронов.
Дополнительные диагностические процедуры
До появления в рутинной клинической практике методов молекулярной диагностики для постановки диагноза СМА использовалось несколько методов исследования, включая электрофизиологические и биопсию мышц. В настоящее время эти методы используются лишь в атипичных случаях и у лиц, отрицательных по делеции в гене SMN1. Методы электрофизиологического обследования, такие как: ЭМГ, исследование электрического вызванного ответа мышцы и оценка количества двигательных единиц, позволяют получить результаты, коррелирующие с особенностями фенотипа заболевания (в частности, двигательными нарушениями), в связи с чем были предложены для мониторинга течения СМА и для оценки исходов при проведении клинических исследований.
В настоящее время проводится разработка дополнительных биомаркеров СМА и надежных способов оценки исходов заболевания, которая позволит более точно оценивать состояние пациентов в клинических исследованиях и судить об эффективности изучаемого терапевтического воздействия.

Сокращения:
СМА – спинальная мышечная атрофия
SMN – (белок) выживаемости мотонейронов
Ссылки:
1. Селивёрстов Ю.А., Клюшников С.А., Иллариошкин С.Н. Спинальные мышечные атрофии: понятие, дифференциальная диагностика, перспективы лечения // Нервные болезни. 2015. №3. URL: https://cyberleninka.ru/article/n/spinalnye-myshechnye -atrofii-ponyatie-differentsialnaya-diagnostika-perspektivy-lecheniya (дата обращения: 31.08.2020).
2. Darras B.T. Spinal muscular atrophies // Pediatr. Clin. North Am. 2015. V. 62. № 3. P. 743–766.
3. Bromberg MB1, Swoboda KJ. Motor unit number estimation in infants and children with spinal muscular atrophy. // Muscle Nerve. 2002, V.25(3) P.445-7.
4. Faravelli I., Nizzardo M., Comi G.P., Corti S. Spinal muscular atrophy – recent therapeutic advances for an old challenge // Nat. Rev. Neurol. 2015. V. 11. № 6. P. 351–359.
5. Wang C.H., Finkel R.S., Bertini E.S.; Participants of the International Conference on SMA Standard of Care. Consensus statement for standard of care in spinal muscular atrophy // J. Child Neurol. 2007. V. 22. № 8. P. 1027–1049
M-RU-00002210 Февраль 2021